
As a chemistry major and a psychiatrist Lithium has a special place in my consciousness. During the years that I took organic chemistry lithium aluminum hydride was a favorite reducing agent when creating certain organic syntheses. Most chemistry majors remember metallic lithium as one of those highly reactive metals that was packed under oil to prevent contact with water or even moisture in the air. Lithium's reactivity is why the free metal does not exist in nature. The most common form used as a medication is lithium carbonate in various preparations.
There has been a lot of speculation about the mechanism of action of lithium since its discovery. Early in my career the definitive source of information for all things lithium-related was the Lithium Information Center at the University of Wisconsin Department of Psychiatry. It was possible to call them and ask them anything about lithium and get the relevant references sent to you. They also produced the Lithium Encyclopedia for Clinical Practice. The mechanism of action was described as unknown at the time but ongoing research was cited (p. 7) in "ion substitution with subsequent effects on amine metabolism, membrane transport, glucose metabolism, and neurotransmitter synthesis and degradation." An entire chapter was dedicated to mechanism of action. In that chapter, the review of what was known about the mechanism of action at the time is interesting. The major neurotransmitter systems being studied at the time were catecholamines, serotonin, and acetylcholine. Animal studies showed acute changes on norepinephrine turnover that was only slight to non-existent with chronic use. Results on serotonin turnover were conflicting, but it prevented hyperaggressive behavior resulting from a serotonin depleting compound that blocked tryptophan hydroxylase (parachlorophenylalanine). Acute administration did not alter dopamine turnover. Chronic administration resulted in increased turnover in mesolimbic and striatal areas but not the cortex. These observations led to theories that lithium worked by altering post synaptic receptor sensitivity including decreased beta adrenoreceptor effects, stabilized opioid receptors, and preventing dopamine receptor hypersensitivity.
There was some speculation about endocrine mechanisms since it was known that lithium blocks release of thyroid hormone (T4). It was also believed to reduce testosterone levels as a possible role in the anti-aggressive properties of the medication. Studies at the time showed that in patients treated for aggression and closely followed, they had increased levels of luteinizing hormone but normal testosterone levels. A significant theory at the time was that lithium worked by reducing T4 levels and this reduced beta-adrenoreceptor potentiation in mood disorders. Lithium was also thought to possibly work by the effect it had on the intracellular concentration of other ions like sodium, calcium, potassium, and magnesium in neurons. The 1980s was a decade when research interest on cell signalling was becoming more widespread after Sutherland's Nobel Prize for the discovery of cyclic AMP (cAMP). Lithium was noted to inhibit adenylate cyclase the enzyme that produces cAMP. Specific forms linked to beta-adrenoreceptors and prostaglandin-E1 were noted to be blocked leading to speculation that these mechanisms were related to mania.
Another definitive source of drug mechanisms over the same era was The Biochemical Basis of Neuropharmacology. My collection of these texts starts in 1984 with the fourth edition of that text. There was a single paragraph on the action of lithium and its effect on catecholamines. They used the term facilitated recapture mechanism (2) suggesting that the overall block of stimulus related norepinephrine (NE) release may be due to facilitated uptake of NE. They also point out the difference in acute and chronic effects with supersensitive NE responses with chronic administration. By the fifth edition of this text (3), the speculative mechanisms had expanded to include inhibiting inositol-1-phosphatase in the phosphoinositide pathway (p. 114), the same NE mechanism as the previous edition (p. 306), and a new observation that lithium facilitates tryptophan uptake initially but with chronic administration tryptophan production normalizes despite increased uptake due to decreased enzymatic conversion to serotonin (5HT). Shifting the balance between synthesis and uptake was suggested as a more stable mechanism. By the seventh edition, lithium was back to being mentioned on single page as part of the larger discussion of deficits in the catecholamine hypothesis of mood disorders - a theme the authors started in the fourth edition. By the eighth and final version of this text there was no mention of lithium at all. Two of the authors were involved in a successive text called Introduction to Neuropsychopharmacology (5). That text describes lithium as "one of the major achievements of psychopharmacology of the past 50 years (p. 321). The authors acknowledge that the mechanism of action remained unclear but the theories included inhibition of inositol monophosphatase, inhibition of glycogen synthase kinase-β, and modulating g protein function (p. 321).
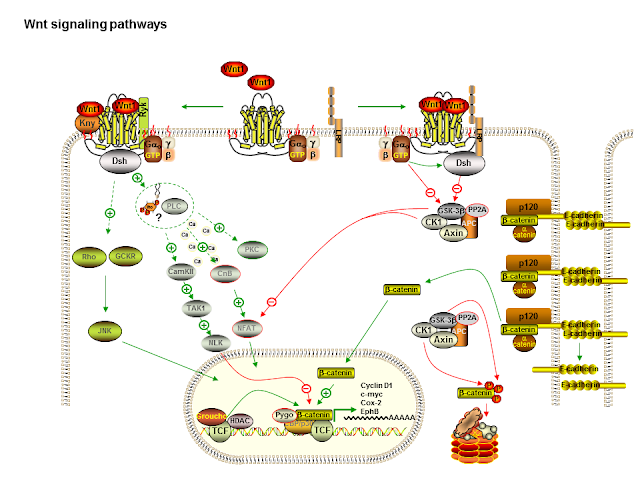
Another excellent source of the evolution of lithium theory was the American College of Neuropsychopharmacology's (ACNP) Generation of Progress series. The series has been discontinued. I have the third to the fifth editions and the most substantial section on lithium action was in the Fifth Generation of Progress (6). This chapter begins with an overview of the time course of mood stabilizer action and how the focus had changed over the previous 20 years from changes in neurotransmitter release and regulation to changes in cell signalling and morphological changes consistent with "altered signalling in critical regions of the brain." The chapter is an overview of the complex effects of lithium on ion transport, neurotransmitter release, signal transduction, circadian rhythm, gene expression, and neuroplasticity. The data showing that lithium and in some cases valproate and carbamazepine can regulate gene expression via transcription factors is reviewed. Some of the changes produced a neuroprotective effect against a number of factors and at about that time neuroprotection was considered a potential positive effect form both mood stabilizer and antidepressants. It was very interesting to reread the section on neuroplasticity. Lithium was known to be an inhibitor of glycogen synthase kinase-3 beta (GSK-3β). This molecule is a component of the wnt signalling pathway (see diagram). This inhibition results in reduced phosphorylation of tau protein and the overall effect of increased microtubule assembly. Phosphorylation of MAP-1β is also inhibited by lithium and this results in increased axonal spreading and increased growth cone area and perimeter. Long term down regulation of protein kinase C (PKC) substrate myrisolated alanine-rich c-kinase substrate (MARCKS) via phosphoinositide signalling was also shown to MARCKS expression is high in developmentally important structures like neuronal growth cones necessary for brain development. It is also high in limbic structures in the human brain that retain the potential for plasticity - like learning and memory. The authors conclude that section by pointing out that by its action on PI/PKC and GSK-3β signalling cascades, lithium "may alter presynaptic and post-synaptic structure to stabilize aberrant neuronal activity in critical areas of the brain involved in the regulation of mood." In the space of just a few years, lithium was suddenly implicated in neuroplasticity and neuroprotection. Maybe the Decade of the Brain did produce some benefits?
That brings me to the latest piece of the puzzle. A paper from Molecular Psychiatry (7) this October further examines the role of these signalling systems and how everything comes together. The authors propose that one common biochemical pathway that may confer susceptibility to psychiatric disorders is the Wnt/ β-catenin pathway. This is a pathway that is critical in all multicellular organisms for cell differentiation, growth, proliferation and morphology across a number of organ systems. At least part of the pathway has a direct influence on the cytoskeleton. It has been implicated in human diseases especially tumors and the metabolism of tumors. The pathway was discovered about 34 years ago. The authors also looked at DIX domain containing-1 (DIXDC1) as a cytoplasmic transducer of the Wnt/ β-catenin pathway. DIXDC1 interacts with disrupted in schizophrenia-1 (DISC-1) gene that has been implicated in the genetics of schizophrenia, bipolar disorder, and autism spectrum disorder. DIXDC1 also has a restricted distribution in the nervous system depending on developmental stages.
Like most modern papers this article has an intense experimental section. The authors prepared a DIXDC1 knockout mouse model and looked at several experimental manipulations. They used several behavioral pharmacology approaches to model anxiety, depression, and social interaction among the mice. On these models the Dixdc1KO (knock out) mice showed increased depression, increased anxiety, and less socialization than the Dixdc1WT (wild type) mice. These behavioral phenotypes correlated with histological changes and the Dixdc1KO mice had reduced spine density and an increased number of filopodial or immature spines on pyramidal cell dendrites. The authors confirmed that these reduced spine neurons functioned in an electrophysiologically expected manner. They analyzed the reduced spines in the Dixdc1KO mutants and found that there was a decreased density of glutamatergic synapses along the dendrites of pyramidal neurons. In order to determine if the Dixdc1KO Wnt/ β-catenin pathway would be impaired by the loss of cytoplasmic signal transduction proteins. They found that treating the KO and WT neurons with and activator (Wnt3a) - the level of β-catenin rose as expected in the WT neurons. Wnt3a also failed to effect spine maturity or glutamatergic synapse density on the KO type neurons.
Most importantly for psychiatrists, the authors hypothesized that lithium would correct both the behavioral phenotype and structural defects in the Dixdc1KO type mice by inhibition of GSK3. Injection of lithium or the specific GSK inhibitor GSK3i corrected the behavioral phenotypes and spine density, spine morphology, and glutamatergic synapse density in the pyramidal neurons of Dixdc1KO mice.
In a separate experiment the authors looked at a large database of patients with psychiatric disorders. The first database contained 6000 cases of autism spectrum disorder (ASD) and 7000 controls. The ASD cases had a greater number of sequence disrupting single-nucleotide variants (SNVs) that were judged to be likely to disrupt DIXDC1 function. They showed the same pattern in patients with bipolar disorder (BD) and schizophrenia (Scz) versus controls. In the end they had 4 patient data sets totaling 9000 cases (versus 11000 controls) with significantly more rare sequence disrupting SNVs.
The authors also used a cell based Wnt/ β-catenin signalling assay (compared to WT) to test specific missense SNVs from both psychiatric patients (BD, Scz) and ASD patients. They found that rare missense SNV from ASD patients either increased or decreased Wnt/ β-catenin pathway activation. Rare missense SNVs from psychiatric patients did not rescue spine density and synaptic deficits but the WT did. A number of Wnt/ β-catenin pathway hyperactivating SNVs cased the expected decreased spine density, decreased glutamatergic synapse density and increased immature spine density.
The authors conclude that there may be other mechanisms in play that they could have missed. They cite a downstream mechanism that is independent of the Wnt/ β-catenin pathway that leads to the structural changes they monitored in this study. There are also different isoforms of DIXDC1 - some more active than others. They do a great job of summarizing 20 years of research in the following lines:
"Several different biochemical mechanisms have been proposed to underlie the anxiolytic, antidepressant and mood stabilizing properties of lithium, a drug whose systemic use in modern psychiatry began in the first half of the last century. Lithium's best validated mechanisms of action are inhibitory on IMP and INPP1, central phosphatases in the phosphoinositide pathway and on GSK3, the central kinase in the Wnt/ β-catenin pathway and AKT pathways." (p. 8).
The story of lithium is similar to a lot of stories in biomedicine. Research on lithium reflects a lot of popular theories of the day rather than any particular unique theory by one scientist. That says a lot about the difference between physical sciences and biological sciences. The technique of applying the most popular theories and lab techniques at the time is still common in medicine and neuroscience. Like most neuroscience there needs to be further testing, replication, and debate about this mechanism but it does seem to be a lot clearer now than at any time in the past. If the mechanism does check out there may be more than the few applications that currently involve lithium. A lithium like effect from safer medications is potentially a very interesting one. Applications may be as diverse as treating addiction - where glutamatergic innervation is thought to be an important component of top down control from the frontal cortex to neurodegenerative disorders and neuroprotection of synaptic complexity.
George Dawson, MD, DFAPA
References:
1: James W. Jefferson, John H. Griest, Deborah L. Ackerman. Lithium Encyclopedia for Clinical Practice. American Psychiatric Press, Washington, DC; 1983.
2: Jack R. Cooper, Floyd E. Bloom, Robert H. Roth. The Biochemical Basis of Neuropharmacology. 4th ed. Oxford, England: Oxford University Press, 1982: 214.
3: Jack R. Cooper, Floyd E. Bloom, Robert H. Roth. The Biochemical Basis of Neuropharmacology. 5th ed. Oxford, England: Oxford University Press, 1985: 115, 306, 319.
4: Jack R. Cooper, Floyd E. Bloom, Robert H. Roth. The Biochemical Basis of Neuropharmacology. 7th ed. Oxford, England: Oxford University Press, 1996: 490.
5: Leslie L. Iverson, Susan D. Iverson, Floyd E. Bloom, Robert H. Roth. Introduction to Neuropsychopharamcology. New York, New York: Oxford University Press, 2009: 321-322.
6: Robert H. Lenox, Alan Frazer. Mechanism of action of antidepressants and mood stabilizers. In: Davis KL, Charney D, Coyle JT, Nemeroff C, eds. Neuropsychopharmacology: The Fifth Generation of Progress. Philadelphia, Pennsylvania: Lippincott, Williams, and Wilkins, 2002: 1139-1163.
7: Martin PM, Stanley RE, Ross AP, Freitas AE, Moyer CE, Brumback AC, Iafrati J, Stapornwongkul KS, Dominguez S, Kivimäe S, Mulligan KA, Pirooznia M, McCombie WR, Potash JB, Zandi PP, Purcell SM, Sanders SJ, Zuo Y, Sohal VS, Cheyette BN. DIXDC1 contributes to psychiatric susceptibility by regulating dendritic spine and glutamatergic synapse density via GSK3 and Wnt/β-catenin signaling. Mol Psychiatry. 2016 Oct 18. doi: 10.1038/mp.2016.184. [Epub ahead of print] PubMed PMID: 27752079.
8: Saito-Diaz K, Chen TW, Wang X, Thorne CA, Wallace HA, Page-McCaw A, Lee E. The way Wnt works: components and mechanism. Growth Factors. 2013 Feb;31(1):1-31. doi: 10.3109/08977194.2012.752737. Review. PubMed PMID: 23256519
There has been a lot of speculation about the mechanism of action of lithium since its discovery. Early in my career the definitive source of information for all things lithium-related was the Lithium Information Center at the University of Wisconsin Department of Psychiatry. It was possible to call them and ask them anything about lithium and get the relevant references sent to you. They also produced the Lithium Encyclopedia for Clinical Practice. The mechanism of action was described as unknown at the time but ongoing research was cited (p. 7) in "ion substitution with subsequent effects on amine metabolism, membrane transport, glucose metabolism, and neurotransmitter synthesis and degradation." An entire chapter was dedicated to mechanism of action. In that chapter, the review of what was known about the mechanism of action at the time is interesting. The major neurotransmitter systems being studied at the time were catecholamines, serotonin, and acetylcholine. Animal studies showed acute changes on norepinephrine turnover that was only slight to non-existent with chronic use. Results on serotonin turnover were conflicting, but it prevented hyperaggressive behavior resulting from a serotonin depleting compound that blocked tryptophan hydroxylase (parachlorophenylalanine). Acute administration did not alter dopamine turnover. Chronic administration resulted in increased turnover in mesolimbic and striatal areas but not the cortex. These observations led to theories that lithium worked by altering post synaptic receptor sensitivity including decreased beta adrenoreceptor effects, stabilized opioid receptors, and preventing dopamine receptor hypersensitivity.
There was some speculation about endocrine mechanisms since it was known that lithium blocks release of thyroid hormone (T4). It was also believed to reduce testosterone levels as a possible role in the anti-aggressive properties of the medication. Studies at the time showed that in patients treated for aggression and closely followed, they had increased levels of luteinizing hormone but normal testosterone levels. A significant theory at the time was that lithium worked by reducing T4 levels and this reduced beta-adrenoreceptor potentiation in mood disorders. Lithium was also thought to possibly work by the effect it had on the intracellular concentration of other ions like sodium, calcium, potassium, and magnesium in neurons. The 1980s was a decade when research interest on cell signalling was becoming more widespread after Sutherland's Nobel Prize for the discovery of cyclic AMP (cAMP). Lithium was noted to inhibit adenylate cyclase the enzyme that produces cAMP. Specific forms linked to beta-adrenoreceptors and prostaglandin-E1 were noted to be blocked leading to speculation that these mechanisms were related to mania.
Another definitive source of drug mechanisms over the same era was The Biochemical Basis of Neuropharmacology. My collection of these texts starts in 1984 with the fourth edition of that text. There was a single paragraph on the action of lithium and its effect on catecholamines. They used the term facilitated recapture mechanism (2) suggesting that the overall block of stimulus related norepinephrine (NE) release may be due to facilitated uptake of NE. They also point out the difference in acute and chronic effects with supersensitive NE responses with chronic administration. By the fifth edition of this text (3), the speculative mechanisms had expanded to include inhibiting inositol-1-phosphatase in the phosphoinositide pathway (p. 114), the same NE mechanism as the previous edition (p. 306), and a new observation that lithium facilitates tryptophan uptake initially but with chronic administration tryptophan production normalizes despite increased uptake due to decreased enzymatic conversion to serotonin (5HT). Shifting the balance between synthesis and uptake was suggested as a more stable mechanism. By the seventh edition, lithium was back to being mentioned on single page as part of the larger discussion of deficits in the catecholamine hypothesis of mood disorders - a theme the authors started in the fourth edition. By the eighth and final version of this text there was no mention of lithium at all. Two of the authors were involved in a successive text called Introduction to Neuropsychopharmacology (5). That text describes lithium as "one of the major achievements of psychopharmacology of the past 50 years (p. 321). The authors acknowledge that the mechanism of action remained unclear but the theories included inhibition of inositol monophosphatase, inhibition of glycogen synthase kinase-β, and modulating g protein function (p. 321).
Another excellent source of the evolution of lithium theory was the American College of Neuropsychopharmacology's (ACNP) Generation of Progress series. The series has been discontinued. I have the third to the fifth editions and the most substantial section on lithium action was in the Fifth Generation of Progress (6). This chapter begins with an overview of the time course of mood stabilizer action and how the focus had changed over the previous 20 years from changes in neurotransmitter release and regulation to changes in cell signalling and morphological changes consistent with "altered signalling in critical regions of the brain." The chapter is an overview of the complex effects of lithium on ion transport, neurotransmitter release, signal transduction, circadian rhythm, gene expression, and neuroplasticity. The data showing that lithium and in some cases valproate and carbamazepine can regulate gene expression via transcription factors is reviewed. Some of the changes produced a neuroprotective effect against a number of factors and at about that time neuroprotection was considered a potential positive effect form both mood stabilizer and antidepressants. It was very interesting to reread the section on neuroplasticity. Lithium was known to be an inhibitor of glycogen synthase kinase-3 beta (GSK-3β). This molecule is a component of the wnt signalling pathway (see diagram). This inhibition results in reduced phosphorylation of tau protein and the overall effect of increased microtubule assembly. Phosphorylation of MAP-1β is also inhibited by lithium and this results in increased axonal spreading and increased growth cone area and perimeter. Long term down regulation of protein kinase C (PKC) substrate myrisolated alanine-rich c-kinase substrate (MARCKS) via phosphoinositide signalling was also shown to MARCKS expression is high in developmentally important structures like neuronal growth cones necessary for brain development. It is also high in limbic structures in the human brain that retain the potential for plasticity - like learning and memory. The authors conclude that section by pointing out that by its action on PI/PKC and GSK-3β signalling cascades, lithium "may alter presynaptic and post-synaptic structure to stabilize aberrant neuronal activity in critical areas of the brain involved in the regulation of mood." In the space of just a few years, lithium was suddenly implicated in neuroplasticity and neuroprotection. Maybe the Decade of the Brain did produce some benefits?
That brings me to the latest piece of the puzzle. A paper from Molecular Psychiatry (7) this October further examines the role of these signalling systems and how everything comes together. The authors propose that one common biochemical pathway that may confer susceptibility to psychiatric disorders is the Wnt/ β-catenin pathway. This is a pathway that is critical in all multicellular organisms for cell differentiation, growth, proliferation and morphology across a number of organ systems. At least part of the pathway has a direct influence on the cytoskeleton. It has been implicated in human diseases especially tumors and the metabolism of tumors. The pathway was discovered about 34 years ago. The authors also looked at DIX domain containing-1 (DIXDC1) as a cytoplasmic transducer of the Wnt/ β-catenin pathway. DIXDC1 interacts with disrupted in schizophrenia-1 (DISC-1) gene that has been implicated in the genetics of schizophrenia, bipolar disorder, and autism spectrum disorder. DIXDC1 also has a restricted distribution in the nervous system depending on developmental stages.
Like most modern papers this article has an intense experimental section. The authors prepared a DIXDC1 knockout mouse model and looked at several experimental manipulations. They used several behavioral pharmacology approaches to model anxiety, depression, and social interaction among the mice. On these models the Dixdc1KO (knock out) mice showed increased depression, increased anxiety, and less socialization than the Dixdc1WT (wild type) mice. These behavioral phenotypes correlated with histological changes and the Dixdc1KO mice had reduced spine density and an increased number of filopodial or immature spines on pyramidal cell dendrites. The authors confirmed that these reduced spine neurons functioned in an electrophysiologically expected manner. They analyzed the reduced spines in the Dixdc1KO mutants and found that there was a decreased density of glutamatergic synapses along the dendrites of pyramidal neurons. In order to determine if the Dixdc1KO Wnt/ β-catenin pathway would be impaired by the loss of cytoplasmic signal transduction proteins. They found that treating the KO and WT neurons with and activator (Wnt3a) - the level of β-catenin rose as expected in the WT neurons. Wnt3a also failed to effect spine maturity or glutamatergic synapse density on the KO type neurons.
Most importantly for psychiatrists, the authors hypothesized that lithium would correct both the behavioral phenotype and structural defects in the Dixdc1KO type mice by inhibition of GSK3. Injection of lithium or the specific GSK inhibitor GSK3i corrected the behavioral phenotypes and spine density, spine morphology, and glutamatergic synapse density in the pyramidal neurons of Dixdc1KO mice.
In a separate experiment the authors looked at a large database of patients with psychiatric disorders. The first database contained 6000 cases of autism spectrum disorder (ASD) and 7000 controls. The ASD cases had a greater number of sequence disrupting single-nucleotide variants (SNVs) that were judged to be likely to disrupt DIXDC1 function. They showed the same pattern in patients with bipolar disorder (BD) and schizophrenia (Scz) versus controls. In the end they had 4 patient data sets totaling 9000 cases (versus 11000 controls) with significantly more rare sequence disrupting SNVs.
The authors also used a cell based Wnt/ β-catenin signalling assay (compared to WT) to test specific missense SNVs from both psychiatric patients (BD, Scz) and ASD patients. They found that rare missense SNV from ASD patients either increased or decreased Wnt/ β-catenin pathway activation. Rare missense SNVs from psychiatric patients did not rescue spine density and synaptic deficits but the WT did. A number of Wnt/ β-catenin pathway hyperactivating SNVs cased the expected decreased spine density, decreased glutamatergic synapse density and increased immature spine density.
The authors conclude that there may be other mechanisms in play that they could have missed. They cite a downstream mechanism that is independent of the Wnt/ β-catenin pathway that leads to the structural changes they monitored in this study. There are also different isoforms of DIXDC1 - some more active than others. They do a great job of summarizing 20 years of research in the following lines:
"Several different biochemical mechanisms have been proposed to underlie the anxiolytic, antidepressant and mood stabilizing properties of lithium, a drug whose systemic use in modern psychiatry began in the first half of the last century. Lithium's best validated mechanisms of action are inhibitory on IMP and INPP1, central phosphatases in the phosphoinositide pathway and on GSK3, the central kinase in the Wnt/ β-catenin pathway and AKT pathways." (p. 8).
The story of lithium is similar to a lot of stories in biomedicine. Research on lithium reflects a lot of popular theories of the day rather than any particular unique theory by one scientist. That says a lot about the difference between physical sciences and biological sciences. The technique of applying the most popular theories and lab techniques at the time is still common in medicine and neuroscience. Like most neuroscience there needs to be further testing, replication, and debate about this mechanism but it does seem to be a lot clearer now than at any time in the past. If the mechanism does check out there may be more than the few applications that currently involve lithium. A lithium like effect from safer medications is potentially a very interesting one. Applications may be as diverse as treating addiction - where glutamatergic innervation is thought to be an important component of top down control from the frontal cortex to neurodegenerative disorders and neuroprotection of synaptic complexity.

George Dawson, MD, DFAPA
References:
1: James W. Jefferson, John H. Griest, Deborah L. Ackerman. Lithium Encyclopedia for Clinical Practice. American Psychiatric Press, Washington, DC; 1983.
2: Jack R. Cooper, Floyd E. Bloom, Robert H. Roth. The Biochemical Basis of Neuropharmacology. 4th ed. Oxford, England: Oxford University Press, 1982: 214.
3: Jack R. Cooper, Floyd E. Bloom, Robert H. Roth. The Biochemical Basis of Neuropharmacology. 5th ed. Oxford, England: Oxford University Press, 1985: 115, 306, 319.
4: Jack R. Cooper, Floyd E. Bloom, Robert H. Roth. The Biochemical Basis of Neuropharmacology. 7th ed. Oxford, England: Oxford University Press, 1996: 490.
5: Leslie L. Iverson, Susan D. Iverson, Floyd E. Bloom, Robert H. Roth. Introduction to Neuropsychopharamcology. New York, New York: Oxford University Press, 2009: 321-322.
6: Robert H. Lenox, Alan Frazer. Mechanism of action of antidepressants and mood stabilizers. In: Davis KL, Charney D, Coyle JT, Nemeroff C, eds. Neuropsychopharmacology: The Fifth Generation of Progress. Philadelphia, Pennsylvania: Lippincott, Williams, and Wilkins, 2002: 1139-1163.
7: Martin PM, Stanley RE, Ross AP, Freitas AE, Moyer CE, Brumback AC, Iafrati J, Stapornwongkul KS, Dominguez S, Kivimäe S, Mulligan KA, Pirooznia M, McCombie WR, Potash JB, Zandi PP, Purcell SM, Sanders SJ, Zuo Y, Sohal VS, Cheyette BN. DIXDC1 contributes to psychiatric susceptibility by regulating dendritic spine and glutamatergic synapse density via GSK3 and Wnt/β-catenin signaling. Mol Psychiatry. 2016 Oct 18. doi: 10.1038/mp.2016.184. [Epub ahead of print] PubMed PMID: 27752079.
8: Saito-Diaz K, Chen TW, Wang X, Thorne CA, Wallace HA, Page-McCaw A, Lee E. The way Wnt works: components and mechanism. Growth Factors. 2013 Feb;31(1):1-31. doi: 10.3109/08977194.2012.752737. Review. PubMed PMID: 23256519
Could the dampening of neuronal excitability by extra blood calcium, resulting from lithium usage, be how lithium works ?
ReplyDeleteProbably not. The mild hypercalcemia associated with chronic lithium use is not a uniform effect and occurs in a minority of patients. It is thought to be caused by increased parathyroid hormone secretion due to the effect of lithium on the feedback loop in which serum calcium inhibits the secretion of parathyroid hormone (PTH). The effect of PTH is to activate osteooclasts that lead to bone resorption and increased intestinal and renal calcium reabsorption and increased calcium levels.
DeletePsychiatric patients may be hypocalcemic and lithium may only need to restore calcium levels to normal for symptoms to clear up.
DeleteThe effect is so uncertain it could not possibly account for clear cut therapeutic effect of lithium. Secondary calcium effects in a few people do not explain the therapeutic effects of lithium.
DeleteI forgot to add that hypocalcemia does not cause mania or depression.
Delete